" Design History File (DHF), Device Master Record (DMR) 和 Device History Record (DHR) 這三者的關聯性為何呢? 在什麼階段需要產出這些文件呢? 這些文件跟醫療器材品質系統又有什麼關聯呢? 若您有上述的疑惑,不妨參考一下本篇的探討!"
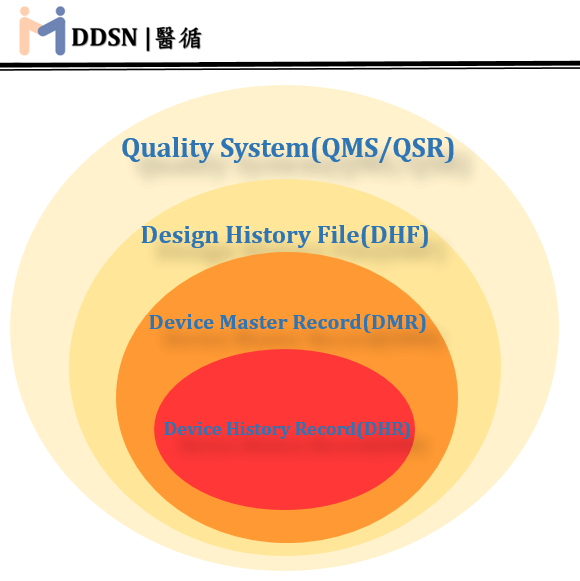
Quality System (QMS/ QSR):
「品質系統泛指設計開發、製造、檢驗、銷售、客訴、公司資源管理…等多項活動,只要會影響醫療器材之生命週期的事件,都需要透過建立品質系統來進行管控.」 由於各國法規機構對於品質系統的要求,都是採取各自頒布相關的條文/標準來規範廠商,雖然有逐漸走向調和各國法規的趨勢,不過目前各國對於品質系統的要求還是有些差異.舉歐美國家為例,大家耳熟能詳的ISO 13485就是歐盟用來規範廠商建立Quality Management System(QMS)之標準;而美國FDA則是頒布21 CFR Part 820 - Quality System Regulation(QSR)來規範廠商!
美國FDA將Quality System劃分為7個Subsystems,其中Design Control就是一個獨立的Subsystem,而且Design Control, Corrective and Preventive Action (CAPA), Management與Production and Process Controls為4個Major Subsystems,由此可看出美國FDA對於Design Control的重視性!(而ISO 13485是將Design Control放在clause 7. 產品實現的章節內)

2. Quantity manufactured
3. Quantity released for distribution
4. Acceptance records which demonstrate the device is manufactured in accordance with DMR
- 21 CFR Part 820 - Quality System Regulation(QSR)
美國FDA將Quality System劃分為7個Subsystems,其中Design Control就是一個獨立的Subsystem,而且Design Control, Corrective and Preventive Action (CAPA), Management與Production and Process Controls為4個Major Subsystems,由此可看出美國FDA對於Design Control的重視性!(而ISO 13485是將Design Control放在clause 7. 產品實現的章節內)
Design History File (DHF):
「DHF指的是設計開發管制流程(Design Control)下所產出的相關文件.簡單來說就是 “如何將無形的客戶/臨床需求轉變成有形的產品規格”以及“如何建立可穩定生產的產品規格”這兩個過程之文件化,其中後者就是指DMR,所以完成了DHF也會產出DMR.」 美國FDA頒布了21 CFR Part 820.30 - Design controls.來說明該如何執行Design Control,而在最後一項(j)即是解釋何為DHF,FDA的定義為 " Design history file. Each manufacturer shall establish and maintain a DHF for each type of device. The DHF shall contain or reference the records necessary to demonstrate that the design was developed in accordance with the approved design plan and the requirements of this part. "
此外,貼心的FDA還繪製了Design Control Flowchart (如下圖),方便廠商了解Design Control的實施方式,從圖中我們也可以了解到,在執行Design Control流程中所產出的相關文件即為DHF.

(Ref. US FDA)
值得留意的是,在ISO 13485:2003 中其實並未提及到DHF,不過到了新版
ISO 13485:2016中,則是新增了 7.3.10 Design and development files條文,其內容為 "The organization shall maintain a design and development file for each medical device type or medical
device family. This file shall include or reference records generated to demonstrate conformity to the
requirements for design and development and records for design and development changes. "
這意味著歐盟開始要求廠商建置DHF,以便管控廠商的設計開發過程,不難觀察出歐美對於DHF的重視性逐漸一致!
延伸閱讀:苦惱Design Control該如何進行嗎?整備Design History File (DHF)的過程遇到阻礙了嗎?不妨參考醫循(MDDSN)所提供的服務!
Device Master Record (DMR):
「DMR是指文件化產品製造生產與檢驗所需的相關作業.透過DMR可讓公司的製造部或是委外的製造廠了解如何生產這項產品.」 在前面所提到的Design Control Flowchart 中,可以看到Design Output即會包含產出DMR,但需要注意的是Design Output涵括各階段Design Control所產出的全部文件,而DMR只有針對最終產品的規格與生產程序紀錄,也就是說DMR只是Design Output的其中一部分.在美國FDA 21 CFR Part 820.3 - Definitions.(j),對於DMR的定義為 "Device master record (DMR ) means a compilation of records containing the procedures and specifications for a finished device." ;而在21 CFR Part 820.181 - Device master record.則是進一步定義了DMR該產出的內容,整理如下:
1. Device specifications
2. Production process specifications
3. Quality assurance procedures and specifications
4. Packaging and labeling specifications
5. Installation, maintenance and servicing procedures and methods
1. Device specifications
2. Production process specifications
3. Quality assurance procedures and specifications
4. Packaging and labeling specifications
5. Installation, maintenance and servicing procedures and methods
Device History Record (DHR):
「DHR指的是每批次生產的歷史紀錄,DHR通常是由製造方所產出.」在美國FDA 21 CFR Part 820.3 - Definitions.(i),對於DHR的定義為 "Device history record (DHR ) means a compilation of records containing the production history of a finished device." ;而在21 CFR Part 820.184-Device history record.則是進一步定義了DHR該產出的內容,整理如下:
1. Dates of manufacture2. Quantity manufactured
3. Quantity released for distribution
4. Acceptance records which demonstrate the device is manufactured in accordance with DMR
討論:
透過本篇文章可以了解到DHF, DMR和DHR這三者之間的關係,簡單來說DHF紀錄了醫材產品從無到有誕生的過程;而DMR則是記錄著如何生產/製造/檢驗這項產品;最後DHR則是每批次的生產紀錄!由於DHF記錄著產品設計的依據以及設計開發管制過程,因此建立完善的DHF不僅能協助廠商快速取得法規上市許可;也能讓公司在每一次設計變更或CACP的過程中都具有可追溯性.引用資料:
- “ MDDSN | 醫循 — 提供醫療器材商品化一條可依循的開發取證途徑! ”- 21 CFR Part 820 - Quality System Regulation(QSR)
留言
張貼留言